ChIP-Atlas 3.0: a data-mining suite to explore chromosome architecture together with large-scale regulome data
Zhaonan Zou, Tazro Ohta, Shinya Oki
Nucleic Acids Research 2024 May 16;gkae358
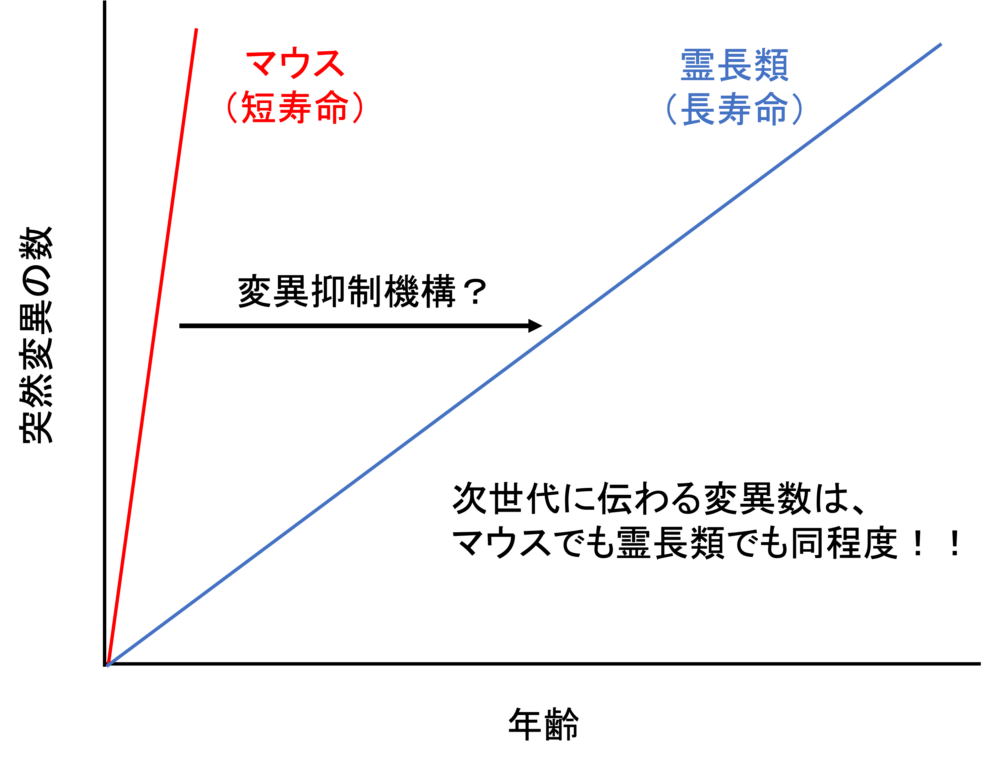
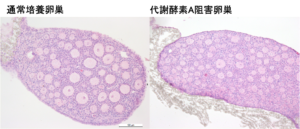
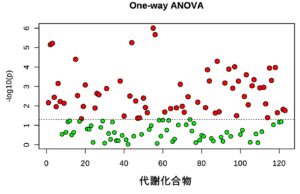
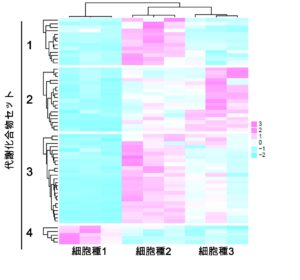
真核生物の遺伝子発現は、細胞特異的なエピゲノム状態やゲノムへのタンパク結合、ヒストン修飾パターンによって時空間的に精密に制御されています。この発現制御機構を理解するため、我々は数年前より ChIP-Atlas(https://chip-atlas.org)というエピゲノミクスの統合データベースを開発してきました。ヒトと5つのモデル生物(マウス、ラット、ショウジョウバエ、線虫、出芽酵母)のゲノム–タンパク質相互作用(ChIP-seq)、オープンクロマチン情報(ATAC-seq)、メチローム情報(Bisulfite-seq)を可視化するとともに、これらをフル活用したデータマイニングツールを公開しています。この度は、ChIP-Atlas のメジャーアップデートを行い、ゲノムの三次元構造(Hi-C)・疾患感受性ゲノム変異(GWAS SNPs)などの注釈づけ情報を統合し、遺伝子発現制御に関わるエピゲノム状態の変容を検出する比較解析ツール Diff Analysis を実装しました。これにより、ChIP-Atlas は高度な解析機能を備えた世界最大規模のデータ数(376,000 実験)を誇るエピゲノム情報インフラへと進化し、遺伝性疾患の発症メカニズムや薬物作用機序の解明、細胞分化転換の研究の効率化などへのさらなる貢献が期待されます。
")